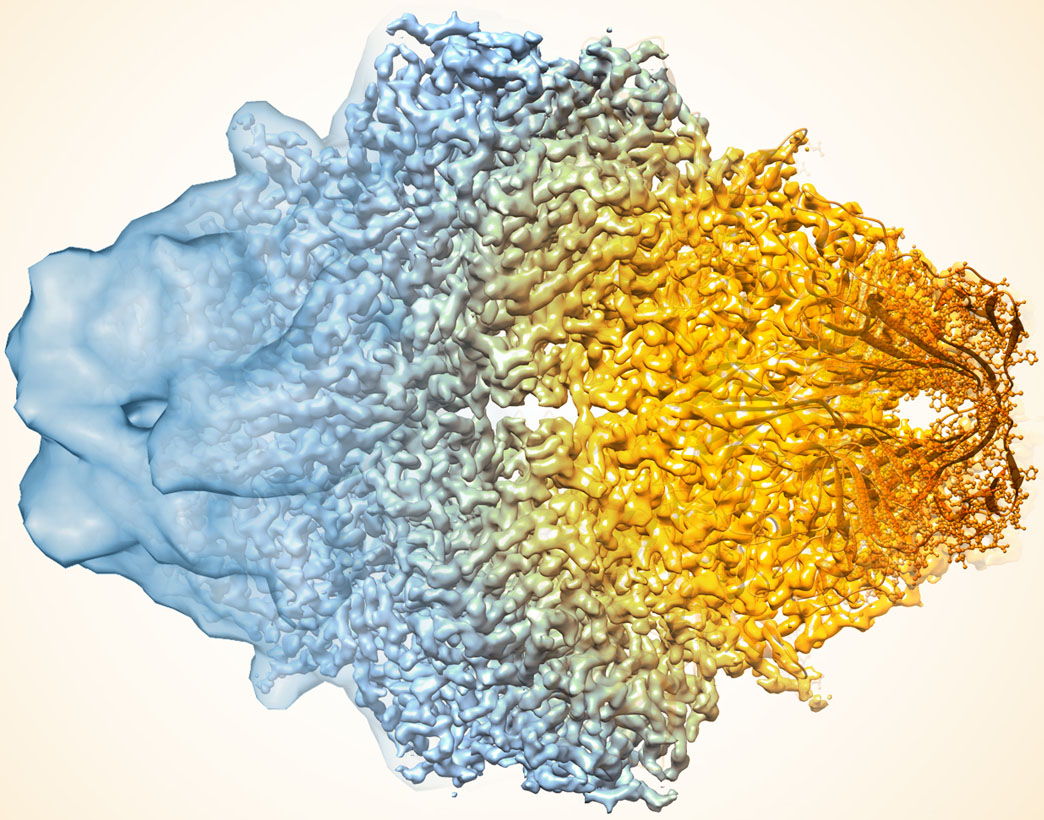
Caption: Composite image of beta-galactosidase showing how cryo-EM’s resolution has improved dramatically in recent years. Older images to the left, more recent to the right.
Credit: Veronica Falconieri, Subramaniam Lab, National Cancer Institute
In the quest to find faster, better ways of mapping the structure of proteins and other key biological molecules, a growing number of researchers are turning to an innovative method that pushes the idea of a freeze frame to a whole new level: cryo-electron microscopy (cryo-EM). The technique, which involves flash-freezing molecules in liquid nitrogen and bombarding them with electrons to capture their images with a special camera, has advanced dramatically since its inception thanks to the efforts of many creative minds. In fact, cryo-EM has improved so much that its mapping performance now rivals that of X-ray crystallography [1], the long-time workhorse of drug developers and structural biologists.
To get an idea of just how far cryo-EM has come over the last decade, take a look at the composite image above, which shows a bacterial enzyme (beta-galactosidase) bound to a drug-like molecule (phenylethyl beta-D-thiogalactopyranoside). To the left, you see a blob-like area generated by cryo-EM methods that would have been considered state-of-the-art just a few years ago. To the right, there’s an exquisitely detailed structure, which was produced at more than 10-times greater resolution using the latest advances in cryo-EM. In fact, today’s cryo-EM is so powerful that researchers can almost make out individual atoms! Very impressive, and just one of the many reasons why the journal Nature Methods recently named cryo-EM its “Method of the Year” for 2015 [2].
The improved resolution owes to steady technological improvement in generating and detecting electrons as well as the emergence of better computational methods to convert thousands of 2D images into a single 3D map. Efforts to reduce image blurring have also been important, as protein samples jiggle around when hit with an electron beam. The direct electron detectors now in use take a series of frames, which are then aligned to produce a higher quality image. By the way, that’s essentially the same way an iPhone takes clear pictures of wiggly kids!
The record-breaking cryo-EM images of beta-galactosidase reported last year in Science by a team at the NIH’s National Cancer Institute, led by Sriram Subramaniam, took advantage of all of these advances and showed that cryo-EM has finally arrived. Although high-resolution cryo-EM images of proteins smaller than the size of antibody molecules haven’t yet been obtained, in principle any protein or protein complex of interest might be fair game. That can’t be said for X-ray crystallography (not all proteins are amenable to crystallization) or nuclear magnetic resonance (NMR) spectroscopy (only small proteins can be resolved).
Already, the number of protein structures solved at high resolution by cryo-EM has risen exponentially, reaching nearly 300 protein maps last year [3]. In addition to the bacterial enzyme, Subramaniam and his colleagues have recently used cryo-EM to explore the structure and function of brain receptors implicated in Alzheimer’s disease and glycoproteins on the surface of HIV.
The implications for drug discovery and development are perhaps most exciting. Cryo-EM maps showing the contacts between small molecules and proteins can help to explore questions such as why one drug is better than another or why certain drugs fail.
These dramatic advances serve as a reminder of the ways in which basic technological innovation can open new realms of scientific possibility. As Robert Glaeser of the Lawrence Berkeley National Laboratory, Berkeley, CA, noted in that Nature Methods issue, these results in cryo-EM, impressive as they are, still fall far short of what physics allows [4]. And that means we can expect those images to keep getting better.
References:
[1] 2.2
{kind=link}